

2025 will mark the 20th anniversary of the Small and Medium Enterprise (SME) regulation, and the European Medicines Agency (EMA) continues to recognise SMEs as important actors in innovation and the development of new medicinal products. Incentives are provided to help SMEs overcome some of the financial and administrative hurdles associated with bringing a product to market.
Do you qualify as a SME?
To benefit from the advantages conferred by SME status, you must first have that status confirmed by the SME office at the EMA. Though the criteria can initially seem clear (<250 employees and either annual turnover ≤€50 million, OR an annual balance-sheet total ≤€43 million), any partner organisations (including investors) may also need to be considered.
If you are struggling to understand if your organisation qualifies for SME status, MAP x AxTalis can help. Our regulatory team has a thorough understanding of the qualification criteria and experience of obtaining and maintaining SME status for European Biotechs.
What are the benefits of SME status?
Access to the SME Office advice on regulatory issues and briefing meetings (more below)
Fee exemptions and reductions for pre- and post-authorisation regulatory procedures, for example 90% reduction in scientific advice fees (100% for designated orphan products). These incentives will remain at the same level after implementation of the new EMA Fee Regulation in 2025.
Be kept up to date with latest developments through SME newsletters and dedicated training events.
The SME Office
EMA’s SME office offers guidance through advising, directing and connecting SMEs with relevant scientific and regulatory contact points within EMA, facilitating medicines development and applications for marketing authorisation. It is possible for SMEs to request a briefing meeting with the SME office, to discuss regulatory issues related to product development, and EMA experts can be consulted if appropriate. However, the opinions given are the personal ones of the experts, are not binding, and a SME briefing meeting is not a substitute for formal scientific advice.
——————————–
Scientific Advice
Through financial incentives the EMA encourages SME developers of Advanced Therapy Medicinal Products (ATMPs) or other innovative products to request scientific advice, from the early stages of development. Though centralised scientific advice is not binding on either sponsor or the EMA, product development that follows this advice is likely to lead to a successful Marketing Authorisation Application (MAA). Research by EMA has shown that, where scientific advice is sought and followed the likelihood of successful MAA is nearly 90% (compared to ~40% if SA is not sought or followed).
How to use EMA scientific advice to support your development
Scientific advice can be used to discuss all aspects of product development with the EMA`s experts (quality, non-clinical, clinical). Importantly, the discussion is prospective in nature, looking at planned development and not an assessment of actual data. Scientific advice is provided by EMA as responses to questions posed by the developer, and the proposed solution to each. Asking the right questions and clearly presenting the company’s position will ensure that the advice received is of maximum benefit to the product development.
Other scientific advice procedures
Though the EMA scientific advice is most relevant when planning for future MAA there are other available routes to scientific advice that can be beneficial to SMEs:
Sponsors can request national scientific advice from a single competent authority. Typically this is done early in development, or prior to conducting a clinical trial in the country concerned.
Simultaneous National Scientific Advice (SNSA) can be requested, though phase 2 of this pilot is scheduled to end this year, it is expected to continue to be possible to request SNSA through 2025. The focus of this advice should be design of multinational clinical trials ahead of submission, ensuring that the protocol aligns with the expectations of the counties that it is to be conducted in.
Scientific Advice Working Party (SAWP)-Clinical Trial Coordination Group (CTCG) pilot, launched in June 2024 as part of the ACT EU initiative. The pilot provides advice on scientific aspects of clinical trials towards clarification of both clinical trial and marketing authorisation. The request should concern a multi-national clinical trial targeting a rare disease, with a complex clinical trial design, regulation issue or other complex dimension.
Pre- CTA advice pilot, also part of the ACT EU initiative, is aimed to discuss regulatory and technical questions prior to submission of a clinical trial application in the EU.
The regulatory team at MAP x AxTalis has significant experience of submitting scientific advice to EMA and National Competent Authorities (NCAs) and can advise SMEs on the best pathway for the stage of their product development.
Latest developments on the clinical trials landscape
31 January 2025 marks the end of the transition period and the full implementation of the EU CTR, the largest change in regulation of clinical trials in 20 years. Since the go-live of the Clinical Trials Information System (CTIS), many of the original issues have been resolved, or work-arounds created:
Registration in EMA’s Organisation management System (OMS) is now only required for sponsors. Removing the requirement for clinical trial sites and other third parties has significantly reduced the administrative burden.
Q-IMPD only applications allow confidential assessment where a third party’s product is used in a clinical trial.
The revised CTIS transparency rules that became applicable on 18 June 2024 and relaunch of the public website, ensure continued transparency for the public, whilst reducing the complexity and workload for CTIS users.
MedEthicsEU created in February 2024 to enhance cooperation between Member States’ Ethics Committees and further support harmonisation in the ethics review of clinical trials in Europe.
All stakeholders are committed to improving the submission portal (CTIS) and removing hurdles to efficient processing of clinical trial applications. Nevertheless, CTIS remains complex and time consuming to use, particularly for SMEs with limited capacity; early accompaniment by an expert can help save both resources and time to approval of your clinical trial.
Urgent reminder, end of the CTR transition period: From end of January 2025 all ongoing clinical trials in the EU must be transitioned to the EU CTR, or the sponsor could face penalties. If your organisation still has a trial ongoing under the clinical trial directive, there is very little time left to transition!
The MAP x AxTalis regulatory team have been involved in implementation of the EU CTR since 2021 and can assist in all aspects of your clinical trial application in the EU.
Medicinal products used in combination with medical devices
The challenges faced by sponsors when conducting clinical trials with medicinal products in combination with medical devices, and the interaction of the CTR, MDR and IVDR have been acknowledged by the EMA. Many of the issues faced have been identified through the COMBINE initiative, and industry looks forward to proposed solutions in 2025, essential to keep the EU as an attractive region to conduct clinical trials.
Implementation of the new HTA legislation
All SMEs should be aware of the new EU Health Technology Assessment Regulation (EU HTAR), which will apply from 12 January 2025, onwards for ATMPs and novel oncology drugs. Across Europe, varying criteria have been used to conduct Health Technology Assessments (HTAs), which can lead to disparities in patient access to healthcare. The objective of the HTA Regulation is to create a more harmonised HTA framework, promoting cooperation among member states and ensuring equitable access to innovative health technologies across Europe.
Understanding the terminology
JCA (Joint Clinical Assessment): Mandatory assessment of clinical relative effectiveness & relative safety
JSC (Joint Scientific Consultation): Optional process to assist developers in preparing for JCA, equivalent to scientific advice
HTD (Health Technology Developer): The applicant for the MAA
PICO (Population, Intervention, Comparator, Outcomes): The assessment scope of the JCA for relative effectiveness, will be defined by the HTA bodies, not the applicant.
What will change under the HTAR?
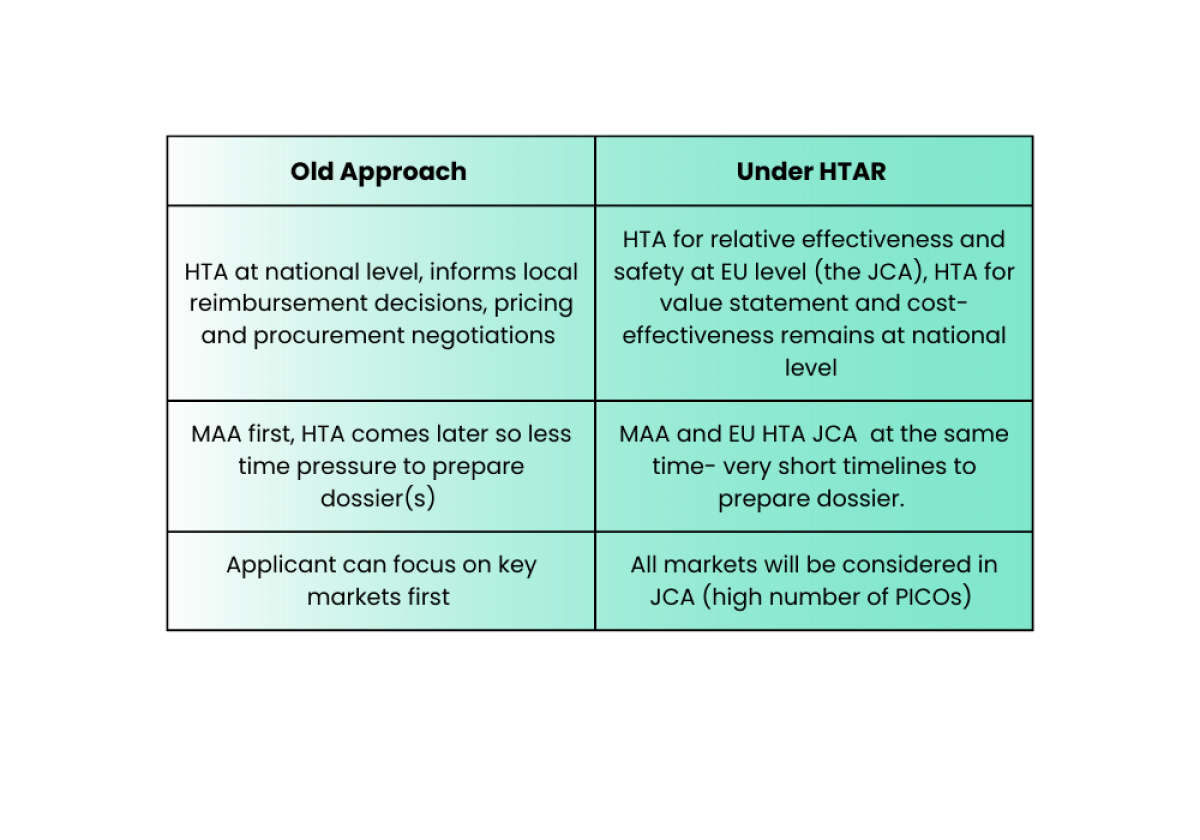
How should SMEs prepare for HTAR?
Though HTA assessment may seem like a distant prospect to a SME with a product in early phases of development, decisions taken then, such as choices of endpoint, could impact the eventual success in market access.
Evidence generation for HTAR should occur earlier in development, in parallel with clinical evidence generation (from Phase I/II)
Important to utilise HTA scientific advice opportunity (JSC), potentially jointly with EMA scientific advice
Proactively consult clinicians and patient advocacy groups, and plan for hypothetical PICOs when designing pivotal studies.
The MAP x AxTalis team have supported over 300 companies with their market access strategy and HTA submissions. Our deep experience means that we have the right tools and expertise to help you overcome both internal and external challenges.
——————————–
SME are a crucial part of the European health ecosystem and are responsible for the discovery and development of many innovative products. However, with limited financial and human resources, it can be difficult for SMEs to navigate the continuously evolving regulatory environment. SME status allows the developer to access financial and administrative incentives, facilitating access to scientific advice, which in turn increases the likelihood of a successful MAA.
The MAP x AxTalis team understand the difficulties that can be faced by SMEs when developing innovative products and can provide solutions customised to your stage of development. To find out how our team can support you in that process, please contact us via email info@axtalis.com.
